
Fluorescence In Situ Hybridization (FISH) protocol
FISH Principle
Fluorescence in situ hybridization (FISH) is a technique that uses fluorescent probes which bind to special sites of the chromosome with a high degree of sequence complementarity to the probes. The fluorescent probes are nucleic acid labeled with fluorescent groups and can bind to specific DNA/RNA sequences. Thus, we can understand where and when a specific DNA sequences exist in cells by detecting the fluorescent group. It was developed in the early 1980s. Fluorescence microscopy can be used to find out where the fluorescent probe is bound to the chromosomes and flow cytometry can be used to detect the binding quantitatively. This FISH protocol is for a Cy5 and FAM labeled probe used in flow cytometry detection and fluorescence microscopy detection.
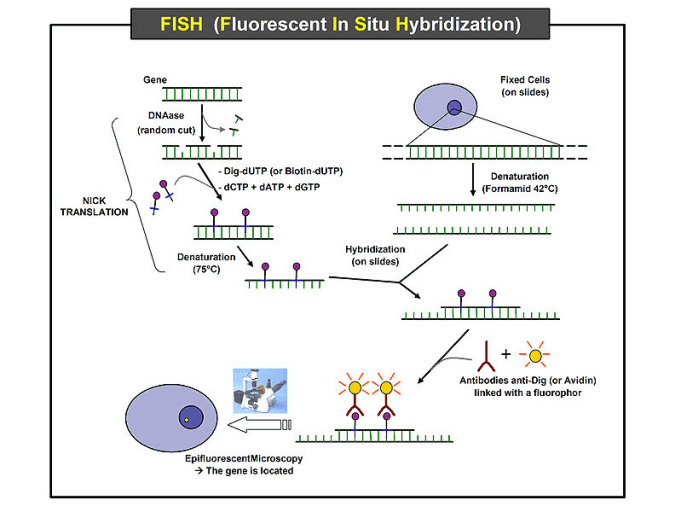
Fig. The schematic diagram of fluorescence in situ hybridization (FISH). The resource come from Wikipedia.
FISH protocol
For flow cytometry detection
- Probes are synthesized and end-labeled at the 5’-end with either Cy5 or 6-carboxyfluorescein (FAM) and purified via HPLC. Cy5-labeled probes are used in experiments involving conventional flow cytometry and FAM-labeled probes are used for experiments involving microscopy or imaging cytometry. The probes is used as a dual probe cocktail at a total probe concentration of 5 ng/µl (2.5 ng/µl each probe).
- Cell samples are pelleted for 5 minutes at 2,000 x and fixed for 30 min at 25°C with 500 µl 10% neutral buffered formalin before FISH procedure.
- After fixation, the formalin is discarded and samples are washed once in 1x PBS.
- Fixed samples are harvested via centrifugation (5 min, 2,000 x g), the supernatant is discarded and cell pellets are resuspended in 0.5 ml of cell storage solution (a 50:50 mix of PBS:absolute ethanol) and then samples can be stored at -20?C until analyzed.
- For each hybridization reaction, 100 µl of fixed samples are pelleted (5 min, 2,000 x g) and resuspended in 100 µl of hybridization buffer (0.7 M NaCl, 0.1 M Tris [pH 8.0], 0.1% SDS, 10 mM EDTA) containing the dual probe cocktail.
- Samples are hybridized for 30 min on a heat block set to 55ºC, followed by a 30 min wash step at the same temperature in 500 µl hybridization buffer without probe.
- Hybridized samples are pelleted and resuspended in a 50:50 mixture of PBS and absolute ethanol, cooled to -20ºC and ship on ice via overnight.
- Once received, samples are placed and held at -20ºC until used, for up to a week. Samples remained liquid under these storage conditions due to the ethanol content.
- Prior to analysis, tubes are vortexed to resuspend cells and break up loosely associated flocs or aggregates.
- 100 uL samples are pelleted via centrifugation (2,000 x g, 5 min).
- Wash samples with 100 µl PBS + 0.5% (w/v) bovine serum albumin (BSA).
- Centrifuge samples again at 2,000 x g, 5 min.
- Resuspend samples in 50 µl PBS + 0.5% BSA prior to running on the flow cytometer.
For fluorescence microscopy detection
- Prior to FISH, tape-bound cells are fixed for 30 min at 25°C by covering the sample contact area with 500 µl 10% neutral buffered formalin.
- After fixation, the formalin is discarded and the tape is washed once in 1x phosphate buffered saline (PBS).
- Then samples are dehydrated in ethanol (a 50, 80 and 95% (v/v) series, exposure for 3 min to 300 µl ethanol at each concentration) prior to hybridization.
- Samples on tapes are hybridized for 15 minutes at 55?C using a moisture-sealed slide incubation chamber. Briefly, 500 µl volumes of hybridization buffer (0.7 M NaCl, 0.1 M Tris [pH 8.0], 0.1% sodium dodecyl sulfate, 10 mM EDTA, containing probe, preheated to 55 ºC) are applied to the surface of the tape and the chamber’s lid is sealed, creating a moist, temperature controlled environment within the chamber.
- After 15 min, the lid is removed and samples are briefly rinsed with probe-free hybridization buffer, preheated to 55ºC.
- Hybridized cells on tapes are counterstained for 10 minutes in the dark with ~30 µl mounting medium containing 1.5 µg ml-1 4',6-diamidino-2-phenylindole (DAPI).
- Then tapes are mounted with a coverslip and examined using a fluorescence microscope.