


Cell Cycle
Immunology Background
Background
The cell cycle, an essential biological process in all living organisms, controls cell growth, DNA replication, and cell division. This intricate mechanism ensures that daughter cells are genetically identical to the parent cell, playing a critical role in development, tissue repair, and maintenance. Its precise regulation is fundamental to preventing diseases such as cancer, which can result from uncontrolled cell division.
The cell cycle consists of two key phases: interphase, where cell growth and DNA replication occur, and mitosis, the division phase. A network of regulatory proteins and checkpoints meticulously controls these processes, ensuring that each stage is properly completed before the next begins. Dysregulation of these controls can lead to excessive cell proliferation, a hallmark of cancer.
Phases of the Cell Cycle
The cell cycle is typically divided into four primary phases: G1 (Gap 1), S (Synthesis), G2 (Gap 2), and M (Mitosis). In addition, some cells can enter G0, a quiescent state in which they remain metabolically active but do not actively divide.
G1 Phase (Gap 1)
As the first stage of interphase, G1 is characterized by cell growth, increased size, and the synthesis of RNA and proteins necessary for subsequent DNA replication and division. Organelle production also occurs during this phase to ensure that daughter cells inherit sufficient cellular components.
S Phase (Synthesis)
During S phase, DNA replication occurs. Each chromosome in the cell's nucleus is duplicated, resulting in two identical sister chromatids for each chromosome. This process relies on enzymes such as DNA polymerases, helicases, and ligases to unwind the DNA double helix and synthesize new complementary strands.
Replication begins simultaneously at multiple origins along the DNA molecule, allowing for rapid genome duplication. Throughout this phase, the cell uses repair mechanisms, including mismatch repair and base excision repair, to correct errors and ensure the fidelity of the genetic information passed on to progeny.
G2 Phase (Gap 2)
G2 is the final phase of interphase during which the cell continues to grow and prepare for mitosis. During this phase, the cell checks the integrity of the replicated DNA and repairs any errors identified during S phase. During G2, the cell also synthesizes proteins necessary for mitotic events, such as those involved in spindle formation, while centrosomes mature to facilitate chromosome segregation. Energy reserves and biosynthetic activity continue to increase to support the upcoming division.
M Phase (Mitosis)
Mitosis (M phase) is the culmination of the cell cycle, where the cell physically divides to form two genetically identical daughter cells. Mitosis consists of several stages: prophase, prometaphase, metaphase, anaphase, and telophase, followed by cytokinesis. Each stage is essential for the proper segregation of chromosomes to the daughter cells.
- Prophase: Chromatin condenses into visible chromosomes, and the nuclear envelope begins to break down.
- Prometaphase: The mitotic spindle, composed of microtubules, forms and attaches to the kinetochores on chromosomes.
- Metaphase: Chromosomes align along the metaphase plate, ensuring that each daughter cell will receive an equal complement of chromosomes.
- Anaphase: Sister chromatids are pulled apart and move toward opposite poles of the cell.
- Telophase: The nuclear envelope reforms around each set of chromosomes, and the chromosomes begin to decondense.
- Cytokinesis: The cell's cytoplasm is divided, resulting in two separate cells, each containing a full complement of organelles and chromosomes.
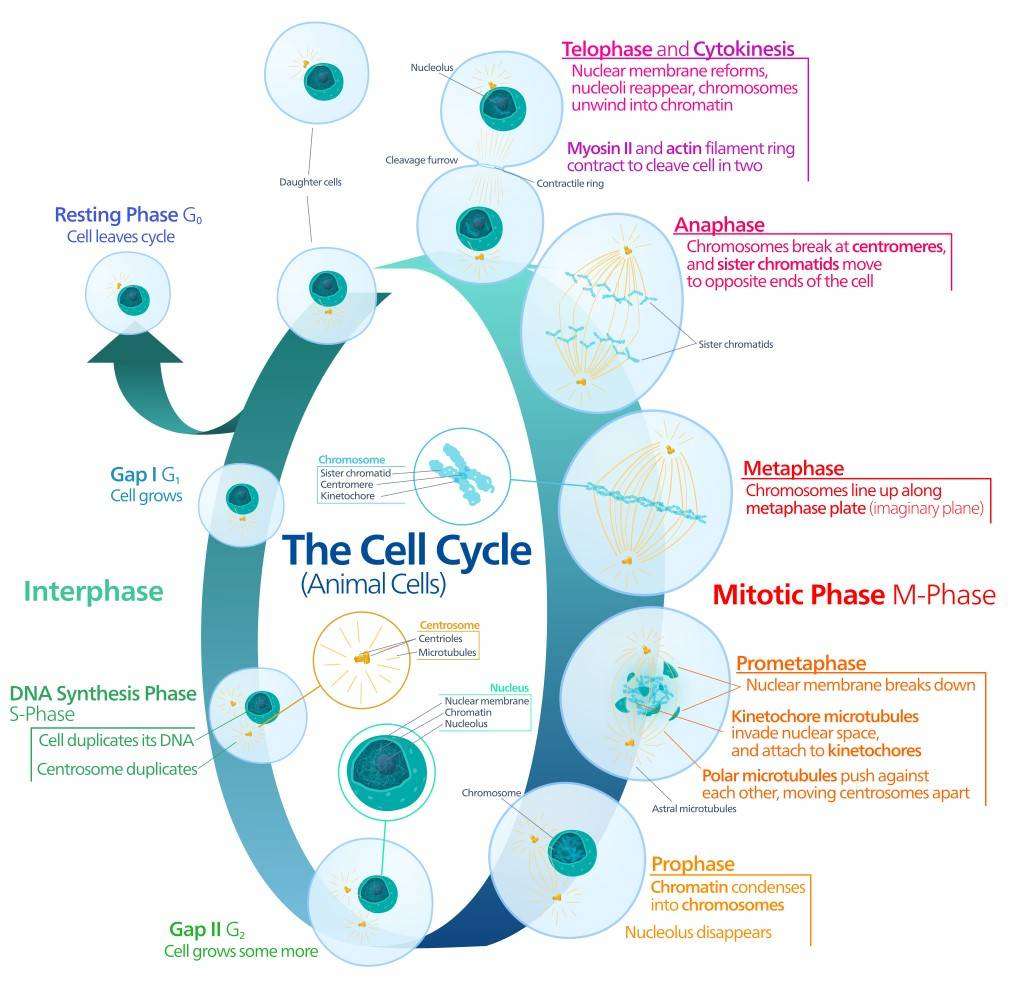 Fig. 1: The key events of animal cell cycle.
Fig. 1: The key events of animal cell cycle.Regulation of the Cell Cycle
The cell cycle is regulated by a complex network of proteins and enzymes that ensure its proper progression. Key regulatory proteins include cyclins, cyclin-dependent kinases (CDKs), and checkpoint proteins, which act as sensors and effectors of cell cycle control.
Cyclins and CDKs: Key Regulators of the Cell Cycle
Cyclins and cyclin-dependent kinases (CDKs) are key regulators of the cell cycle, forming complexes that control the transition between phases. CDKs, a group of serine/threonine kinases, require binding to specific cyclins to become functionally active. Each phase of the cell cycle is controlled by the activation of specific cyclin-CDK complexes, ensuring sequential and controlled progression. While cyclins fluctuate in abundance throughout the cycle to modulate CDK activity, CDKs are maintained at steady levels and are activated only upon cyclin binding.
Cyclin D-CDK4/6: The G1 Phase and G1/S Transition
The G1/S transition is the first major checkpoint that ensures the cell is ready to replicate its DNA. Cyclin D binds to CDK4 or CDK6 to form a complex that phosphorylates retinoblastoma protein (Rb). This phosphorylation releases E2F transcription factors (e.g., E2F1, E2F2, E2F4), which initiate transcription of genes necessary for DNA synthesis.
Cyclin E-CDK2: Commitment to DNA Replication
Cyclin E-CDK2 plays a critical role in the transition from G1 to S phase. Once the cyclin D-CDK4/6 complex initiates Rb phosphorylation, the cyclin E-CDK2 complex completes the process, completely inactivating Rb and driving the cell into S phase. Cyclin E (e.g. CCNE1, CCNE2) levels peak in late G1 and decline after the onset of replication.
Cyclin A-CDK2: Control of S Phase Progression
Upon entry into S phase, cyclin A-CDK2 becomes active to ensure accurate DNA replication. Cyclin A (e.g. cyclin A1, cyclin A2) levels increase in late G1 and associates with CDK2 as the cell enters S phase. The cyclin A-CDK2 complex ensures that DNA replication occurs only once per cell cycle by phosphorylating proteins involved in the initiation and progression of DNA replication. It also prevents re-replication by inhibiting the formation of new replication origins.
Cyclin B-CDK1: G2/M Transition and Mitosis
Cyclin B (e.g., cyclin B1, cyclin B2, cyclin B3), in association with CDK1, drives the cell through the G2/M transition into mitosis. This complex regulates processes such as nuclear envelope disassembly, chromosome condensation, and mitotic spindle assembly. Its activity is controlled by phosphorylation/dephosphorylation events, with activating phosphatases such as Cdc25 enabling mitosis once preparatory processes are complete. Cyclin B degradation after mitosis leads to the inactivation of CDK1, which ensures an orderly exit from the cell cycle.
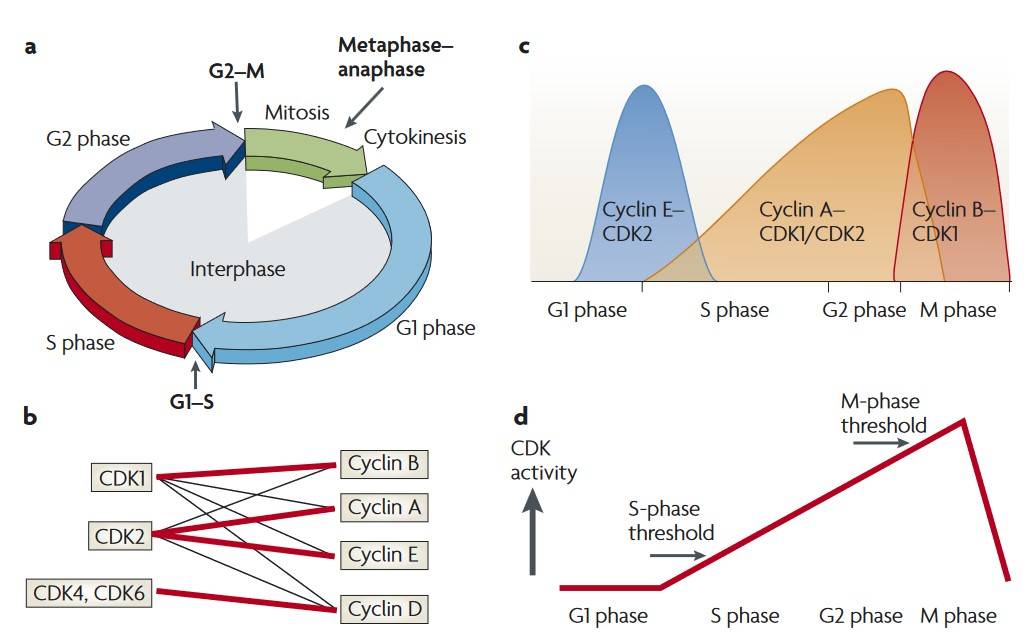 Fig. 2: The classical and minimal models of cell cycle control. a: Phases and checkpoints of the cell cycle. b: CDKs and their partner cyclins. c; According to the classical model of cell cycle control, the levels of the CDK-cyclin complex. D. CDK activity during the cell cycle (Hochegger et al., 2008).
Fig. 2: The classical and minimal models of cell cycle control. a: Phases and checkpoints of the cell cycle. b: CDKs and their partner cyclins. c; According to the classical model of cell cycle control, the levels of the CDK-cyclin complex. D. CDK activity during the cell cycle (Hochegger et al., 2008).Cell Cycle Checkpoints: Ensuring Accuracy and Fidelity
Checkpoints are critical surveillance mechanisms that ensure the proper execution of the cell cycle. This is achieved through monitoring for DNA damage, incomplete replication, and chromosome misalignment. The major checkpoints include the G1/S checkpoint, the G2/M checkpoint, and the spindle assembly checkpoint (SAC).
G1/S Checkpoint: Guarding the Gate to DNA Replication
The G1/S checkpoint ensures that the cell has sufficient energy, nutrients, and growth signals to continue and that the DNA is free of damage. The tumor suppressor p53 plays a critical role in regulating the G1/S checkpoint. When DNA damage is detected, p53 is stabilized and activated, leading to the transcription of p21, a CDK inhibitor that blocks the activity of the cyclin E-CDK2 and cyclin D-CDK4/6 complexes. This results in cell cycle arrest, allowing time for DNA repair. If the damage is too severe to repair, p53 can induce apoptosis to eliminate the damaged cell. Mutations in the p53 gene that disable its function are found in more than 50% of human cancers, underscoring the importance of this checkpoint in preventing malignant transformation.
G2/M Checkpoint: Ensuring DNA Integrity Before Mitosis
The G2/M checkpoint ensures DNA replication completion and repair of any damage before mitosis. This checkpoint is regulated by the ATM/ATR kinases, which detect DNA damage or replication stress and activate checkpoint kinases (Chk1 and Chk2). These kinases inhibit the activity of Cdc25, thereby inhibiting cyclin B-CDK1 activation and ultimately delaying the onset of mitosis. If the damage is repaired, Cdc25 is activated and the cell enters mitosis. However, if the damage cannot be repaired, the G2/M checkpoint can trigger apoptosis, similar to the G1/S checkpoint. Failure of the G2/M checkpoint can lead to the proliferation of damaged or incompletely replicated DNA, contributing to genomic instability and tumorigenesis.
Spindle Assembly Checkpoint (SAC): Preventing Chromosome Mis-Segregation
The spindle assembly checkpoint (SAC) is a critical regulator of mitosis that ensures that all chromosomes are properly attached to the spindle microtubules before anaphase begins. This checkpoint prevents unequal distribution of chromosomes between daughter cells due to premature separation of sister chromatids. This condition is known as aneuploidy. The SAC monitors tension at kinetochores, the protein structures on chromosomes where spindle fibers attach. When the SAC senses improper attachment or insufficient tension, it takes action. It inhibits the anaphase-promoting complex/cyclosome (APC/C), a ubiquitin ligase responsible for degrading key mitotic proteins. In this way, the SAC arrests mitosis to prevent errors in chromosome segregation. Once all chromosomes are properly aligned and attached, the SAC is inactivated. This allows the APC/C to initiate its function, which promotes the degradation of securin and cyclin B. As a result, sister chromatids separate and mitosis proceeds to completion. Defects in SAC components can lead to chromosomal instability, a hallmark of many cancers, as cells with abnormal chromosome numbers can continue to divide and propagate the error.
Integration of Checkpoints and Cancer Development
Checkpoints are essential for preventing genetic mutations that could lead to cancer. These checkpoints act as safeguards, controlling cell division and ensuring genetic integrity. However, when mutations occur in key regulators like p53, Rb, or ATM/ATR, cells can bypass these controls. This leads to the accumulation of mutations that promote tumorigenesis. Many cancers show defects in checkpoint pathways, allowing cells to proliferate despite the presence of DNA damage or chromosomal abnormalities. Research into the molecular mechanisms behind these checkpoints has paved the way for targeted therapies. These include checkpoint kinase inhibitors and drugs designed to restore p53 function or activate the DNA damage response (DDR) in cancer cells.
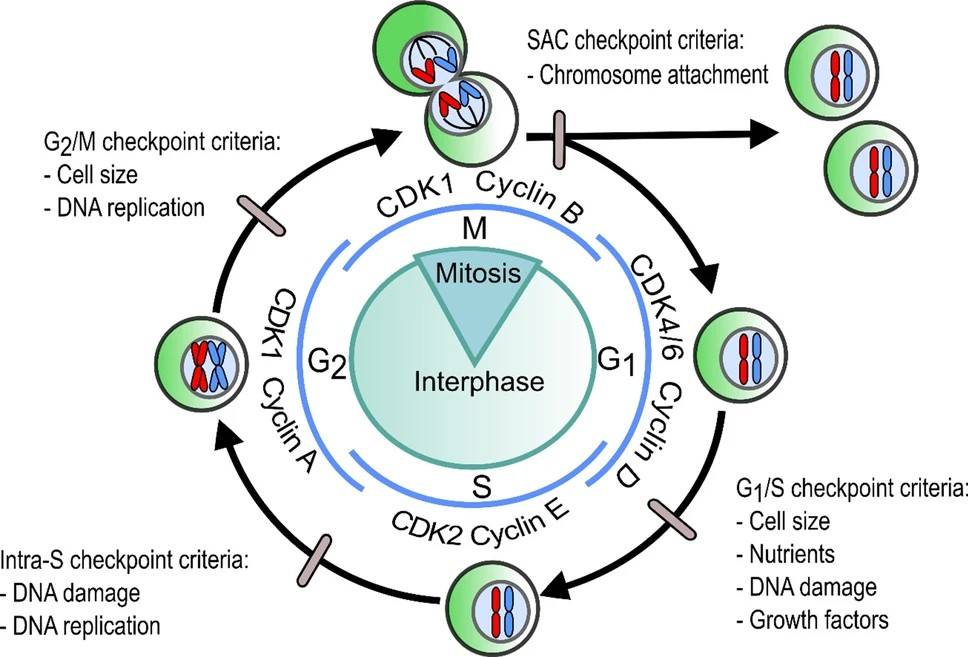 Fig. 3: The generalized cell cycle in eumetazoans illustrating the cell-cycle phase and regulatory checkpoints (grey ovals) with their complementary criteria that must be met for the cell cycle to progress through that checkpoint. Blue lines depict the formation of cell-cycle phase associated CDK-cyclin complexes in Homo sapiens. SAC: Spindle Assembly Checkpoint (Gorman et al., 2020).
Fig. 3: The generalized cell cycle in eumetazoans illustrating the cell-cycle phase and regulatory checkpoints (grey ovals) with their complementary criteria that must be met for the cell cycle to progress through that checkpoint. Blue lines depict the formation of cell-cycle phase associated CDK-cyclin complexes in Homo sapiens. SAC: Spindle Assembly Checkpoint (Gorman et al., 2020).Association with Cancer
Cell cycle regulation is tightly controlled by a balance of pro- and anti-proliferative signals. Mutations in genes that regulate the cell cycle are key drivers in the development of cancer. Two major categories of genes are commonly implicated in cancer: oncogenes and tumor suppressor genes. Oncogenes drive cell proliferation, while tumor suppressor genes inhibit cell division or induce cell death in response to cellular stress or DNA damage. Disruptions in the normal function of either class of genes can lead to uncontrolled cell division and the development of cancer.
Targeting the Cell Cycle in Cancer Therapy
The central role of cell cycle dysregulation in cancer has led to the development of several therapeutic strategies. Traditional chemotherapies, such as DNA-damaging agents (e.g., cisplatin), antimetabolites (e.g., 5-fluorouracil), and microtubule inhibitors (e.g., paclitaxel), target rapidly dividing cells. These treatments disrupt DNA replication, or mitosis, and preferentially kill cancer cells. However, they also affect normally proliferating cells, causing side effects such as hair loss, nausea, and immunosuppression.
In recent years, targeted therapies have been developed to inhibit dysregulated cyclin-CDK complexes. CDK4/6 inhibitors, including palbociclib, ribociclib, and abemaciclib, are approved for hormone receptor-positive breast cancer. These drugs prevent Rb phosphorylation, arresting cells in G1 phase and blocking progression to S phase. By targeting cancer cells with dysregulated cyclin D-CDK4/6 activity, these therapies offer a more precise approach with fewer side effects compared to traditional chemotherapy.
Emerging treatments aim to restore tumor suppressor functions, such as those of p53. While direct targeting of mutant p53 has proven challenging, researchers are developing drugs that reactivate wild-type p53 or exploit vulnerabilities in cancer cells with defective p53. In addition, therapies that target the DNA damage response, such as PARP inhibitors, are effective in cancers with impaired DNA repair mechanisms.
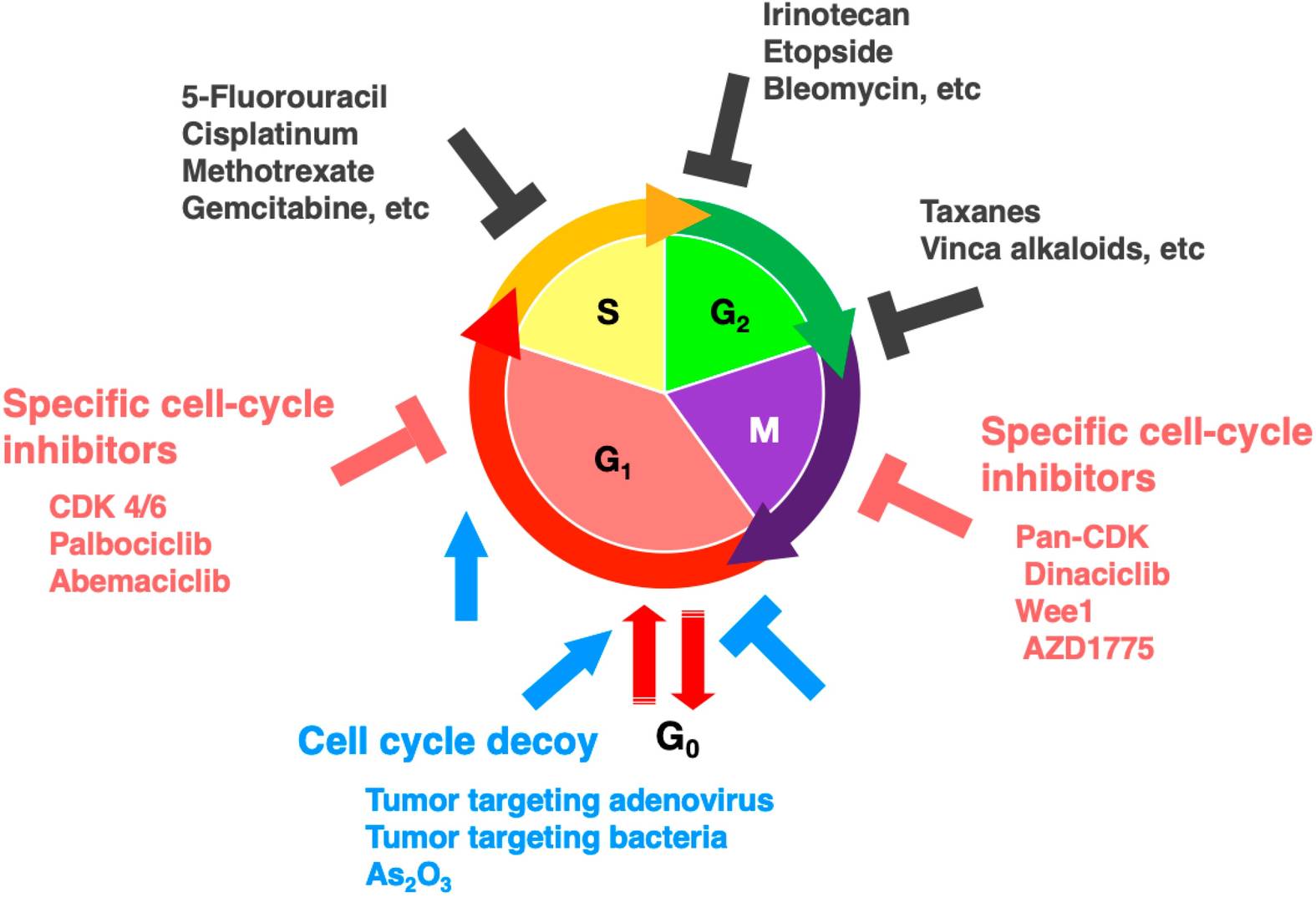 Fig. 4: Scheme of cell-cycle phases where therapeutic agents are most effective.
Fig. 4: Scheme of cell-cycle phases where therapeutic agents are most effective.In summary, the cell cycle is a tightly regulated process that is essential for cell growth, replication, and division. This precise control ensures that cells divide precisely, with errors in DNA replication or chromosome segregation corrected prior to division. When this regulation fails, as in cancer, uncontrolled cell proliferation can occur, driving tumor development and progression. Understanding the molecular mechanisms of cell cycle regulation has led to major breakthroughs in cancer treatment. Many therapies now target specific phases of the cell cycle or exploit the unique vulnerabilities of cancer cells. Ongoing research continues to unravel the complexities of cell cycle regulation and its relationship to cancer. These efforts hold great promise for the development of more targeted and effective treatments in the future.
Case Study
Case 1: Bianco, S.; et al. LRH-1 controls proliferation in breast tumor cells by regulating CDKN1A gene expression. Oncogene. 2015, 34(34), 4509–4518.
Liver receptor homolog-1 (LRH-1, NR5A2) is an orphan nuclear receptor that plays a crucial role in cancer progression, particularly in breast cancer. Researchers discovered that inhibiting LRH-1 impacts the proliferation of various breast cancer cell types, including estrogen receptor α (ERα)-positive and triple-negative (ER-/PR-/HER2-) cells. This effect is associated with the upregulation of the cyclin-dependent kinase inhibitor CDKN1A, independent of p53. Additionally, they found that LRH-1 works in conjunction with FOXA1, binding directly to the CDKN1A promoter and a regulatory region located 62 kb upstream of its transcriptional start site, thereby repressing CDKN1A transcription.
To further explore the molecular mechanism of LRH-1, the researchers used lentiviral short hairpin RNA (shRNA) to knock down LRH-1 expression in a panel of breast cancer cell lines, including ERα-positive MCF7 cells, their antiestrogen-resistant variants LCC2 (tamoxifen-resistant) and LCC9 (tamoxifen/fulvestrant-resistant), MCF7 cells overexpressing HER2 (MCF7/HER2), and triple-negative MDA-MB-231 and BT-549 cells. Significant reductions in MCF7, LCC2 and LCC9 cell numbers were observed when treated with shLRH-1 nos 2 and 3 compared to control shRNA (shCTL). These results demonstrate that LRH-1 regulates both 2D and 3D proliferation of breast cancer cells.
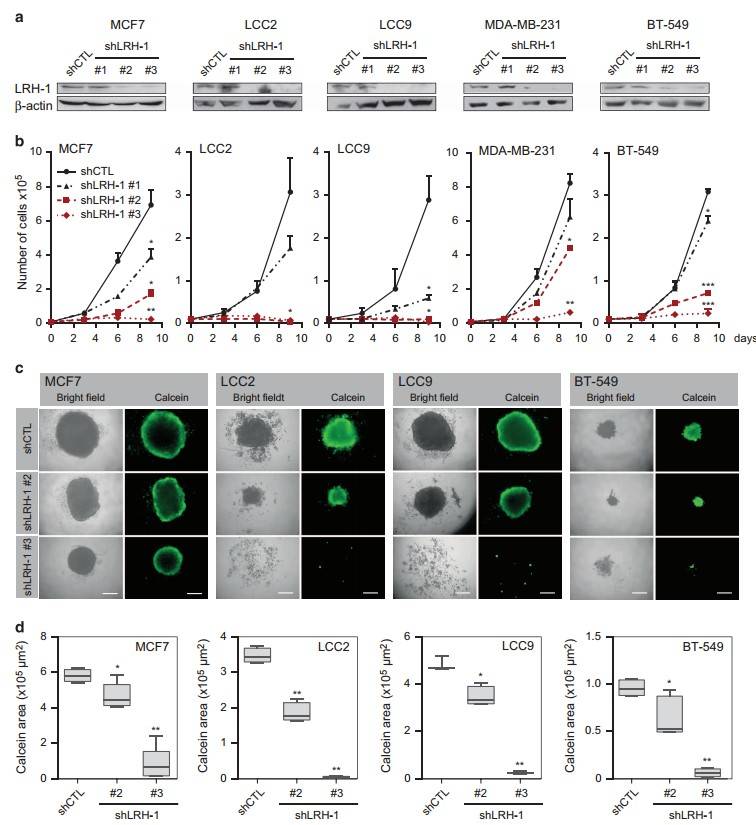 Fig. 5: LRH-1 depletion decreases 2D and 3D cell proliferation of ERα-positive and ERα-negative breast cancer cells. (a) Protein levels of LRH-1 following knockdown analyzed by western blotting. β-Actin was used as a loading control. (b) 2D proliferation assays for cells transduced with shCTL or shLRH-1 lentiviral constructs during 9 days posttransduction (*P<0.05; **P<0.01; ***P<0.001, t-test). (c) Representative bright field and fluorescence images of 3D spheroid cultures of MCF7, LCC2, LCC9 and BT-549 cell lines following LRH-1 knockdown. Viable cells were stained with calcein AM. (d) Spheroid size analysis was performed by measuring calcein area obtained via fluorescence microscopy. Scale bars, 200 μm (*P<0.05; **P<0.001, t-test).
Fig. 5: LRH-1 depletion decreases 2D and 3D cell proliferation of ERα-positive and ERα-negative breast cancer cells. (a) Protein levels of LRH-1 following knockdown analyzed by western blotting. β-Actin was used as a loading control. (b) 2D proliferation assays for cells transduced with shCTL or shLRH-1 lentiviral constructs during 9 days posttransduction (*P<0.05; **P<0.01; ***P<0.001, t-test). (c) Representative bright field and fluorescence images of 3D spheroid cultures of MCF7, LCC2, LCC9 and BT-549 cell lines following LRH-1 knockdown. Viable cells were stained with calcein AM. (d) Spheroid size analysis was performed by measuring calcein area obtained via fluorescence microscopy. Scale bars, 200 μm (*P<0.05; **P<0.001, t-test).To investigate how LRH-1 depletion affects cell proliferation, the Bianco group performed a cell cycle distribution analysis using fluorescence-activated cell sorting. Unexpectedly, they found that LRH-1-depleted breast cancer cells exhibited a strong senescence-associated phenotype. This was characterized by cell cycle arrest, morphological changes such as enlarged and flattened cell shapes, increased β-galactosidase activity, and the formation of heterochromatin foci.
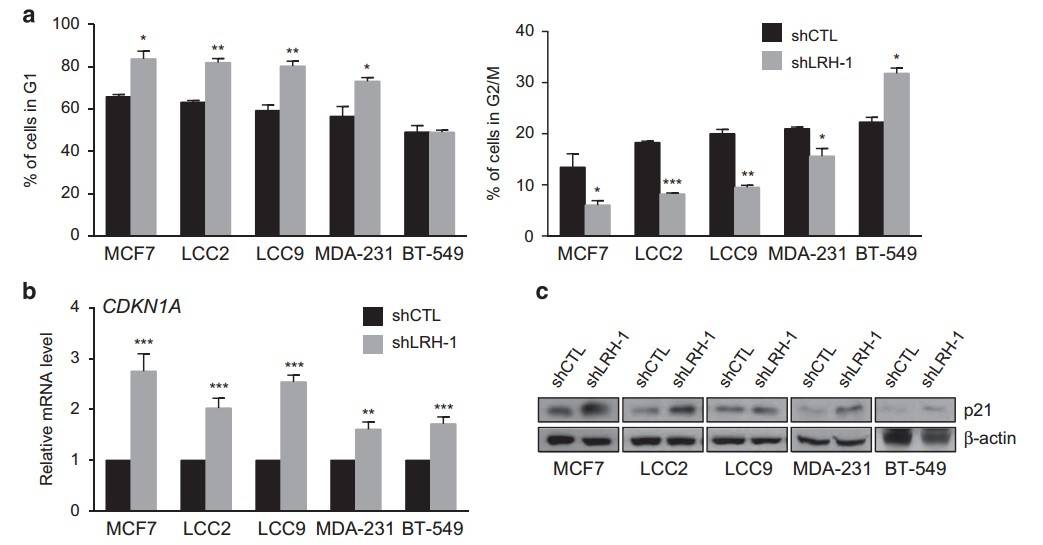 Fig. 6: LRH-1 depletion induces G1- or G2-phase growth arrest and CDKN1 A upregulation. (a) Cell-cycle analysis by fluorescent-activated cell sorting (FACS) showing the number of cells in the G1 (upper panel) and G2/M phase (lower panel) after 4 days of LRH-1 knockdown in MCF7, LCC2, LCC9, MDA-MB-231 (MDA-231) and BT-549 cells. The data shown are the mean ± s.d. of at least three independent experiments. (b) Relative expression of CDKN1A obtained by qPCR following LRH-1 knockdown in different types of breast cancer cells. mRNA expression was normalized relative to the expression level of the housekeeping gene 36B4 and was expressed as the relative expression for shCTL. The data shown are the mean ± s.e.m. of at least three independent experiments (*P<0.05; **P<0.01; ***P<0.001, t-test). (c) Protein levels of CDKN1A/p21 following LRH-1 knockdown were analyzed by western blotting. β-Actin was used as a loading control.
Fig. 6: LRH-1 depletion induces G1- or G2-phase growth arrest and CDKN1 A upregulation. (a) Cell-cycle analysis by fluorescent-activated cell sorting (FACS) showing the number of cells in the G1 (upper panel) and G2/M phase (lower panel) after 4 days of LRH-1 knockdown in MCF7, LCC2, LCC9, MDA-MB-231 (MDA-231) and BT-549 cells. The data shown are the mean ± s.d. of at least three independent experiments. (b) Relative expression of CDKN1A obtained by qPCR following LRH-1 knockdown in different types of breast cancer cells. mRNA expression was normalized relative to the expression level of the housekeeping gene 36B4 and was expressed as the relative expression for shCTL. The data shown are the mean ± s.e.m. of at least three independent experiments (*P<0.05; **P<0.01; ***P<0.001, t-test). (c) Protein levels of CDKN1A/p21 following LRH-1 knockdown were analyzed by western blotting. β-Actin was used as a loading control.Case 2: Wan, B.; et al. AURKB: A promising biomarker in clear cell renal cell carcinoma. PeerJ. 2019, 7, e7718.
Aurora kinase B (AURKB) is a key oncogenic factor in several tumors, but its role in clear cell renal cell carcinoma (ccRCC) remains uncertain. The aim of this study was to evaluate its prognostic significance and mechanism of action in ccRCC. The results showed that AURKB was more highly expressed in ccRCC tissues compared to normal kidney tissues. Kaplan-Meier survival analysis showed that patients with high AURKB expression had a worse prognosis than those with low expression levels.
A comparison of AURKB expression in 539 ccRCC tissues and 72 normal kidney tissues using the Wilcoxon signed-rank test confirmed that AURKB expression was significantly higher in ccRCC. Further analysis of 72 paired samples of ccRCC tissues and adjacent non-cancerous tissues also revealed significant AURKB overexpression in ccRCC. These findings suggest that AURKB may function as an oncogenic gene in ccRCC.
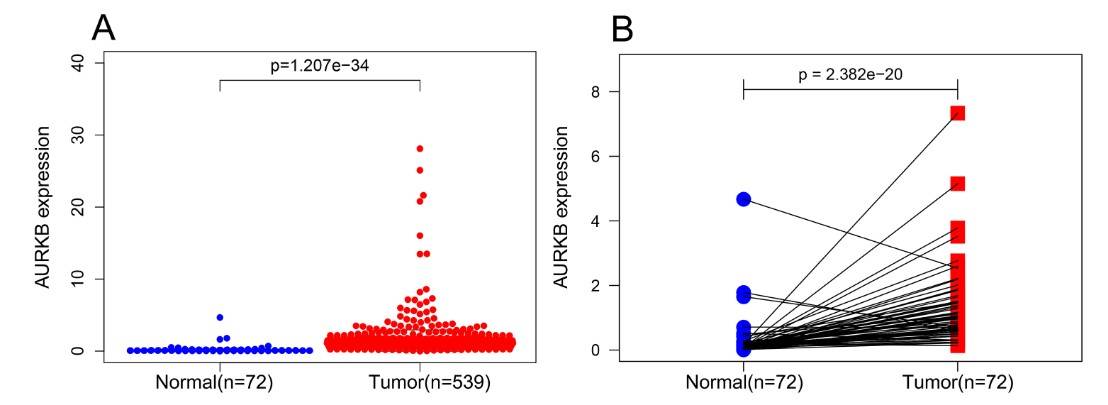 Fig. 7: AURKB was significantly overexpressed in ccRCC compared to normal or adjacent normal tissues. (A) AURKB was significantly upregulated in cancer tissues compared to normal tissues (p < 0.05). (B) AURKB was expressed at higher levels in ccRCC (p < 0.05) compared to 72 pairs of non-cancerous adjacent tissues.
Fig. 7: AURKB was significantly overexpressed in ccRCC compared to normal or adjacent normal tissues. (A) AURKB was significantly upregulated in cancer tissues compared to normal tissues (p < 0.05). (B) AURKB was expressed at higher levels in ccRCC (p < 0.05) compared to 72 pairs of non-cancerous adjacent tissues.To further investigate the prognostic role of AURKB, ccRCC patients were divided into two groups based on the median AURKB expression level (high and low expression groups). Patients with incomplete clinical information were excluded. Kaplan-Meier survival analysis showed that the high AURKB expression group had a worse prognosis than the low expression group. Univariate analysis revealed that high AURKB expression was associated with worse overall survival (OS) and disease-free survival (DFS), and other clinical factors such as pathologic stage and histologic grade were also associated with worse OS and DFS.
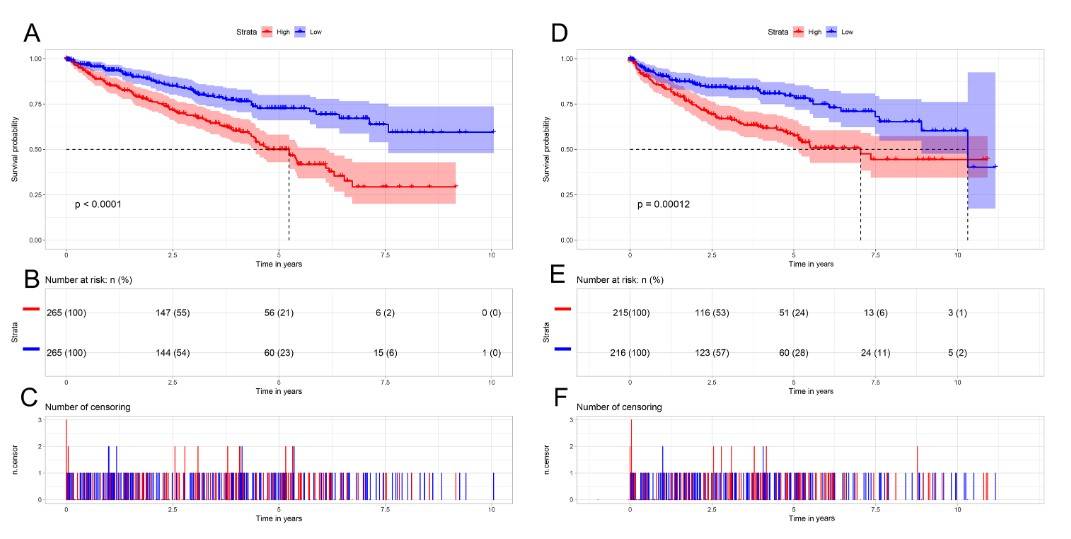 Fig. 8: Kaplan-Meier survival curves of patients with ccRCC based on AURKB expression levels. (A) The Kaplan-Meier curves, (B) number at risk, and (C) number of censoring of OS in ccRCC. (D) The Kaplan-Meier curves, (E) number at risk, and (F) number of censoring of DFS in ccRCC. High expression of AURKB was correlated with poorer OS and DFS in ccRCC patients.
Fig. 8: Kaplan-Meier survival curves of patients with ccRCC based on AURKB expression levels. (A) The Kaplan-Meier curves, (B) number at risk, and (C) number of censoring of OS in ccRCC. (D) The Kaplan-Meier curves, (E) number at risk, and (F) number of censoring of DFS in ccRCC. High expression of AURKB was correlated with poorer OS and DFS in ccRCC patients.References
- Bianco, S., Jangal, M., Garneau, D., & Gévry, N. (2015). LRH-1 controls proliferation in breast tumor cells by regulating CDKN1A gene expression. Oncogene, 34(34), 4509–4518.
- Chen, H., Liu, H., & Qing, G. (2018). Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduction and Targeted Therapy, 3(1), 1–7.
- Freeman-Cook, K., Hoffman, R. L., Miller, N., Almaden, J., Chionis, J., Zhang, Q., Eisele, K., Liu, C., Zhang, C., Huser, N., Nguyen, L., Costa-Jones, C., Niessen, S., Carelli, J., Lapek, J., Weinrich, S. L., Wei, P., McMillan, E., Wilson, E., … Dann, S. G. (2021). Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell,39(10), 1404-1421.e11.
- Gorman, L. M., Wilkinson, S. P., Kitchen, S. A., Oakley, C. A., Grossman, A. R., Weis, V. M., & Davy, S. K. (2020). Phylogenetic analysis of cell-cycle regulatory proteins within the Symbiodiniaceae. Scientific Reports, 10(1), 20473.
- Hochegger, H., Takeda, S., & Hunt, T. (2008). Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nature Reviews Molecular Cell Biology, 9(11), 910–916.
- Knudsen, K. E. (2006). The cyclin D1b splice variant: An old oncogene learns new tricks. Cell Division, 1(1), 15.
- Moulder, D. E., Hatoum, D., Tay, E., Lin, Y., & McGowan, E. M. (2018). The roles of p53 in mitochondrial dynamics and cancer metabolism: The pendulum between survival and death in breast cancer? Cancers, 10(6), 189.
- Spoerri, L., Oo, Z. Y., Larsen, J. E., Haass, N. K., Gabrielli, B., & Pavey, S. (2015). Cell cycle checkpoint and DNA damage response defects as anticancer targets: From molecular mechanisms to therapeutic opportunities. In G. T. Wondrak (Ed.), Stress Response Pathways in Cancer (pp. 29–49). Springer Netherlands.
- Tian, H., Gao, Z., Li, H., Zhang, B., Wang, G., Zhang, Q., Pei, D., & Zheng, J. (2015). DNA damage response – A double-edged sword in cancer prevention and cancer therapy. Cancer Letters, 358(1), 8–16.
- Wan, B., Huang, Y., Liu, B., Lu, L., & Lv, C. (2019). AURKB: A promising biomarker in clear cell renal cell carcinoma. PeerJ, 7, e7718.