
FGF Ligands
Related Symbol Search List
Immunology Background
About FGF Ligands
Fibroblast growth factor (FGF) is a broad-spectrum mitogen that acts as a peptide ligand in a paracrine or endocrine manner.FGF stimulates or maintains specific cellular functions required for metabolism, tissue homeostasis, and development through a signaling axis mediated by the fibroblast growth factor receptor (FGFR). The mammalian FGF family comprises 22 members, all of which share a core segment of approximately 140 amino acids. Based on amino acid sequence and structural analyses, 18 of these members have been classified into 5 paracrine-acting groups (FGF1, FGF4, FGF7, FGF8, and FGF9 subfamilies) and 1 endocrine-acting group (FGF19 subfamily), in addition to an intracellular non-secretory-acting group (FGF11 subfamily).
The binding of FGF ligands to FGFR is localized to the D2 and D3 structural domains of the extracellular domain. The binding of FGFs to inactive monomeric FGFRs triggers conformational changes in FGFRs. Phosphorylating tyrosine residues within the cytoplasmic tails of FGFRs leads to the dimerization and activation of cytoplasmic tyrosine kinases. Functional abnormalities of the FGF & FGFR signaling axis have been observed in various human diseases, such as congenital premature closure of the cranial sutures and dwarfism, as well as chronic kidney disease (CKD), obesity, insulin resistance, and various tumors. The abnormalities are mainly mediated by dysregulated expression of paracrine FGF family proteins and genetic alterations of FGF, FGFR, or cofactor genes, such as mutations, single nucleotide polymorphisms (SNPs), or fusions.
FGF ligands regulate a variety of cellular biological processes, such as cell proliferation, differentiation, and survival, by binding to FGF receptors on the cell surface. In hematopoietic stem cells, FGF ligands are involved in the regulation of HSC proliferation, differentiation, and self-renewal and are essential for maintaining a healthy HSC population in the bone marrow.
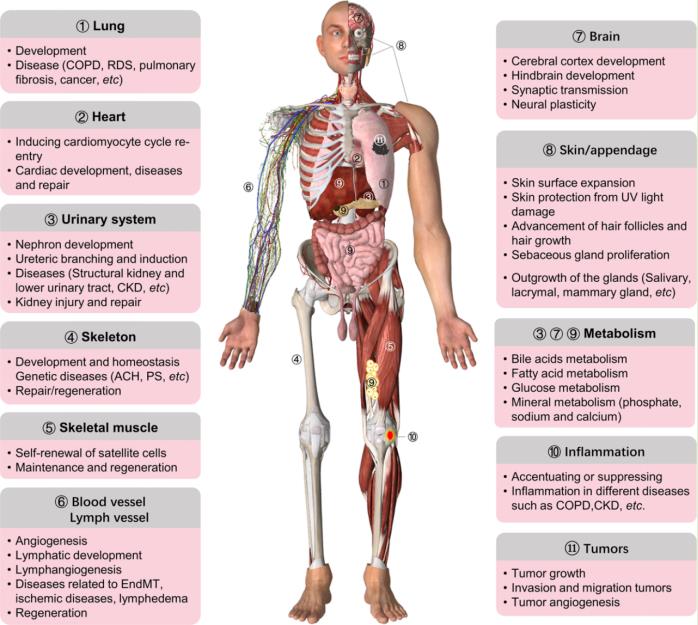 Fig.1 Summary of the main roles of FGF/FGFR signaling in organ development, metabolism, and disease. FGF/FGFR signaling participates in the development of almost all organs such as the lung, heart, urinary system, brain, skeleton, muscle, and skin/appendage, as well as angiogenesis and lymphangiogenesis. FGFs/FGFRs also have important effects on tissue repair, regeneration, and inflammation. Furthermore, endocrine FGFs play critical roles in metabolism by regulating kidney, liver, brain, intestine, and adipose tissue. The malfunctions of FGF/FGFR signaling lead to multiple kinds of diseases, such as genetic diseases, cancer, COPD, and CKD. (Xie Y, et al., 2020)
Fig.1 Summary of the main roles of FGF/FGFR signaling in organ development, metabolism, and disease. FGF/FGFR signaling participates in the development of almost all organs such as the lung, heart, urinary system, brain, skeleton, muscle, and skin/appendage, as well as angiogenesis and lymphangiogenesis. FGFs/FGFRs also have important effects on tissue repair, regeneration, and inflammation. Furthermore, endocrine FGFs play critical roles in metabolism by regulating kidney, liver, brain, intestine, and adipose tissue. The malfunctions of FGF/FGFR signaling lead to multiple kinds of diseases, such as genetic diseases, cancer, COPD, and CKD. (Xie Y, et al., 2020)
Available Products and Information of FGF Ligands
We offer products related to the FGF ligand family, including FGF1, FGF10, FGF11, FGF12, FGF16, FGF17, FGF19, FGF20, FGF21, FGF22, FGF23, FGF4, FGF5, FGF6, FGF7, FGF9. Product types include native proteins, recombinant proteins, cell & tissue lysates, GMP proteins, and protein pre-coupled magnetic meads. Please see the specific product list page for details.
We provide researchers with a range of FGF-related products, as well as detailed descriptions of the products. In addition, you can access important resources such as protein functions, interacting proteins, related articles, related research areas, related signaling pathways, etc., which will provide you with strong support for conducting related research.
Creative BioMart's R&D team has more than a decade of experience in recombinant protein research and development, focusing on innovative protein production and cGMP manufacturing services, as well as a range of related products such as natural proteins, cell/tissue lysates, chromatography, lectins, and assay kits. If you have any questions or need further information about our products, services, or resources, please feel free to contact our team. We are ready to support you and work with you to advance your research and applications.
Reference:
- Xie Y, Su N, Yang J, et al. FGF/FGFR signaling in health and disease[J]. Signal Transduction and Targeted Therapy, 2020, 5(1):181.DOI:10.1038/s41392-020-00222-7.