
Adaptor Proteins in the Akt Pathway
Related Symbol Search List
- CD2AP
- CIDEA
- CRKL
- FADD
- FRS2
- GAB2
- GAPDH
- GRB2
- KHDRBS1
- LAT
- LAT2
- MYD88
- NOD1
- PHF11
- PXN
- 14-3-3 sigma
- SH2B1
- SAP
- SH2D1A
- SHC1
- SOCS5
- SOCS6
- SWAP70
- TRADD
- 14-3-3 beta
- 14-3-3 epsilon
- YWHAG
- YWHAH
- YWHAQ
- YWHAZ
Immunology Background
About Adaptor Proteins in the Akt Pathway
The Akt pathway, also known as the PI3K-Akt pathway, is a crucial cellular signaling pathway involved in various physiological processes, including cell survival, growth, metabolism, and proliferation. This pathway is regulated by a complex network of proteins, including adaptor proteins that play essential roles in transmitting signals from cell surface receptors to downstream effectors. Adaptor proteins serve as molecular bridges, facilitating protein-protein interactions and coordinating the activation and regulation of signaling cascades within the Akt pathway.
The Akt Pathway Overview
The Akt pathway is primarily initiated by the activation of cell surface receptors, such as receptor tyrosine kinases (RTKs) and G-protein coupled receptors (GPCRs). Upon ligand binding, these receptors trigger the activation of phosphatidylinositol 3-kinase (PI3K), which phosphorylates the plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 serves as a critical second messenger that recruits several signaling molecules, including Akt (also known as protein kinase B), to the plasma membrane.
Once recruited to the plasma membrane, Akt is phosphorylated and activated by phosphoinositide-dependent kinase 1 (PDK1) and the mammalian target of rapamycin complex 2 (mTORC2). Activated Akt then phosphorylates a wide range of downstream targets, including proteins involved in cell survival (e.g., Bad and Forkhead box O transcription factors), protein synthesis (e.g., tuberous sclerosis complex 2), and glucose metabolism (e.g., glycogen synthase kinase 3).
Adaptor Proteins in the Akt Pathway
Adaptor proteins play critical roles in the Akt pathway by facilitating protein-protein interactions and promoting the assembly of signaling complexes. These proteins connect the activated cell surface receptors to downstream effectors, enabling efficient signal transmission and amplification. Here are some key adaptor proteins involved in the Akt pathway:
Insulin Receptor Substrate (IRS): IRS proteins are major adaptor proteins in the Akt pathway, particularly in response to insulin and insulin-like growth factor 1 (IGF-1) signaling. IRS proteins are phosphorylated on specific tyrosine residues upon activation of insulin or IGF-1 receptors. Phosphorylated IRS recruits and activates PI3K, initiating the generation of PIP3 and subsequent Akt activation.
Growth Factor Receptor-Bound Protein 2 (Grb2): Grb2 is an adaptor protein that plays a role in linking RTKs to downstream signaling pathways, including the Akt pathway. Grb2 contains Src homology 2 (SH2) domains that bind to phosphorylated tyrosine residues on activated RTKs. Through its association with RTKs, Grb2 can recruit and activate other signaling molecules, such as Son of Sevenless (SOS), which further propagates the signal downstream.
Gab Adaptor Proteins: Gab proteins, including Gab1, Gab2, and Gab3, are adaptor proteins that act downstream of RTKs to transmit signals to various signaling pathways, including the Akt pathway. Gab proteins possess multiple protein-protein interaction domains, such as pleckstrin homology (PH) and Src homology 2 (SH2) domains, which enable their recruitment to phosphorylated tyrosine residues on activated RTKs. Gab proteins then serve as scaffolds to recruit and activate downstream effectors, including PI3K, leading to Akt activation.
Phosphoinositide 3-Kinase Regulatory Subunit (p85): The p85 regulatory subunit of PI3K serves as an adaptor protein in the Akt pathway. It binds to the activated RTKs or other upstream signaling molecules through its SH2 domains, effectively recruiting the catalytic subunit of PI3K (p110) to the plasma membrane. Through this interaction, p85 facilitates the activation of PI3K, leading to the generation of PIP3 and subsequent Akt activation.
These adaptor proteins, along with other components of the Akt pathway, form a complex signaling network that regulates various cellular processes. They enable efficient signal transduction from cell surface receptors to Akt and its downstream effectors, ensuring appropriate cellular responses to external cues.
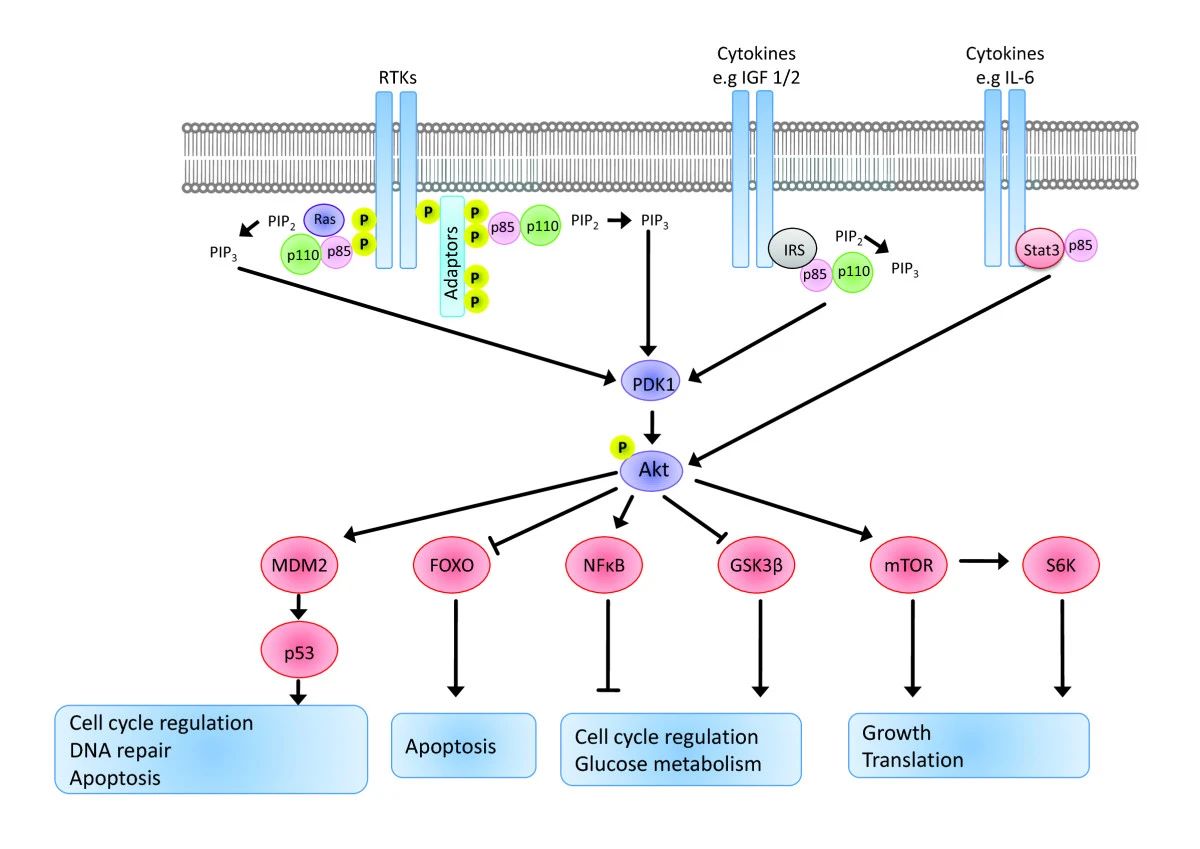 Fig.1 Signalling from receptor tyrosine kinases to Akt is mediated by PI3kinase regulatory subunits and adaptor molecules that activate PI3K, which in turn converts PIP 2 to PIP 3. (Wickenden JA, et al., 2010)
Fig.1 Signalling from receptor tyrosine kinases to Akt is mediated by PI3kinase regulatory subunits and adaptor molecules that activate PI3K, which in turn converts PIP 2 to PIP 3. (Wickenden JA, et al., 2010)
Biological Functions of Adaptor Proteins in the Akt Pathway
Signal Transduction: Adaptor proteins facilitate the transduction of signals from receptor tyrosine kinases (RTKs) or G-protein coupled receptors (GPCRs) to activate Akt signaling. For example, the adaptor protein Grb2 interacts with RTKs and recruits the guanine nucleotide exchange factor SOS, leading to the activation of Ras and subsequent activation of Akt.
Protein-Protein Interactions: Adaptor proteins can mediate specific protein-protein interactions within the Akt pathway. For instance, the protein IRS-1 interacts with the regulatory subunit of phosphoinositide 3-kinase (PI3K) through its PTB domain, leading to the activation of Akt. Additionally, the scaffold protein Gab1 binds to both RTKs and regulatory components of PI3K and facilitates the activation of Akt.
Activation of Akt: Adaptor proteins are vital for the activation of Akt itself. The adaptor protein PDK1 is crucial for the activation of Akt by phosphorylating it at the T-loop residue. PDK1 recognizes the pleckstrin homology (PH) domain of Akt and phosphorylates it, leading to the subsequent activation of Akt.
Integration of Multiple Signaling Pathways: Adaptor proteins help in integrating multiple signaling pathways into the Akt pathway. These proteins can interact with various signaling components and relay signals from different pathways to Akt. For example, the adaptor protein Shc interacts with activated RTKs and subsequently activates Akt signaling by recruiting the PI3K complex.
Applications of Adaptor Proteins in the Akt Pathway
Adaptor proteins in the Akt pathway have important applications in both research and therapeutic settings. Their roles in signal transduction and modulating Akt activation make them valuable targets for studying cellular processes and developing potential interventions. Here are some applications of adaptor proteins in the Akt pathway:
Signal Transduction Studies: Adaptor proteins play a crucial role in transmitting signals from cell surface receptors to downstream effectors in the Akt pathway. Studying the interactions and functions of adaptor proteins provides insights into the mechanisms of signal transduction and regulation within the pathway. Researchers can manipulate the expression or function of adaptor proteins to investigate their specific roles, identify novel protein-protein interactions, and understand how aberrations in these interactions contribute to disease development.
Drug Discovery and Targeting: Adaptor proteins in the Akt pathway represent potential targets for therapeutic interventions. By understanding the roles of adaptor proteins in Akt activation and downstream signaling, researchers can develop strategies to modulate their functions or interactions. Small molecules or biologics can be designed to disrupt specific protein-protein interactions involving adaptor proteins, thereby inhibiting or enhancing Akt pathway signaling. Targeting adaptor proteins may have therapeutic implications for diseases characterized by dysregulated Akt pathway activity, such as cancer, metabolic disorders, and neurodegenerative diseases.
Cancer Therapy: Dysregulation of the Akt pathway is commonly observed in cancer, where it promotes cell survival, proliferation, and resistance to therapy. Adaptor proteins, such as IRS proteins and Gab proteins, are frequently involved in aberrant Akt pathway activation in cancer cells. Targeting these adaptor proteins or their interactions could serve as a therapeutic strategy to inhibit Akt signaling and selectively induce cancer cell death. Modulating adaptor protein function may sensitize cancer cells to conventional therapies or overcome resistance mechanisms, improving treatment outcomes.
Biomarker Identification: Adaptor proteins in the Akt pathway can serve as potential biomarkers for disease diagnosis, prognosis, and therapeutic response prediction. Aberrant expression or activation of adaptor proteins may correlate with specific disease states or treatment outcomes. By analyzing the levels or post-translational modifications of adaptor proteins in patient samples, clinicians and researchers can gain insights into disease progression, identify potential therapeutic targets, and tailor treatment strategies for improved patient management.
Personalized Medicine: The Akt pathway and its associated adaptor proteins exhibit inter-individual variability, influencing responses to therapeutic interventions. Adaptor protein profiles and genetic variants may contribute to differences in Akt pathway activity and treatment outcomes among patients. Integrating information on adaptor protein expression, genetic variations, and Akt pathway status can aid in personalized medicine approaches. This allows for the identification of patient subgroups that may benefit from specific targeted therapies or combination treatments, optimizing treatment efficacy and minimizing adverse effects.
Cellular Engineering and Synthetic Biology: Adaptor proteins can be utilized in synthetic biology approaches to engineer cellular signaling pathways. By manipulating the interactions and functions of adaptor proteins, researchers can rewire signaling networks to achieve desired cellular responses. This can include building artificial signal transduction cascades or designing novel biosensors to detect specific signals or conditions within cells. Adaptor proteins in the Akt pathway can serve as key components in such synthetic biology applications.
In summary, adaptor proteins in the Akt pathway have diverse applications in research and therapeutics. They are valuable for studying signal transduction, identifying therapeutic targets, and developing interventions for diseases characterized by dysregulated Akt pathway activity. Adaptor proteins also hold the potential for biomarker identification, personalized medicine, and engineering cellular signaling networks. Continued research on adaptor proteins and their roles in the Akt pathway will contribute to our understanding of cellular processes and aid in the development of innovative approaches for disease management and treatment.
Available Resources for Adaptor Proteins in the Akt Pathway
Creative BioMart provides products and services covering multiple fields related to protein adapters in the Akt pathway. Products include recombinant proteins, cell and tissue lysates, protein pre-coupled magnetic beads, etc. These products can provide researchers with high-quality experimental tools, helping to deepen our understanding of the role of protein adapters in the Akt pathway, as well as the study and treatment of related diseases.
The following are related molecules/targets of adapter proteins in the Akt pathway. Click on the molecule/target to view research reagents. If you have any questions or requests, please feel free to contact us.
Reference:
- Wickenden JA, Watson CJ. Key signaling nodes in mammary gland development and cancer. Signaling downstream of PI3 kinase in mammary epithelium: a play in 3 Akts. Breast Cancer Res. 2010;12(2):202.