



Recombinant Human PTPN11, GST-tagged, Active 
| Cat.No. : | PTPN11-461H |
| Product Overview : | Recombinant human PTPN11 (246-593) was expressed inE.colicells using a N-terminal GST tag. MW = 69 kDa. |
| Availability | July 18, 2026 |
| Unit | |
| Price | |
| Qty |
- Specification
- Gene Information
- Related Products
- Citation
- Download
| Species : | Human |
| Source : | E.coli |
| Tag : | GST |
| Protein Length : | 246-593 a.a. |
| Description : | Mammalian PTPases can be subdivided into 1 of 2 broad categories: transmembrane receptor PTPases and intracellular PTPases. PTPN11 is one of the 2 closely related mammalian intracellular PTPases whose sequences encode 2 tandem SRC homology 2 (SH2) domains that are located at the amino-terminal side of a single PTPase catalytic domain. This PTP is widely expressed in most tissues and plays a regulatory role in various cell signaling events that are important for a diversity of cell functions, such as mitogenic activation, metabolic control, transcription regulation, and cell migration. |
| Sequence : | 246-593. |
| Applications : | Phosphatase Assay, Western Blot. |
| Storage And Stability : | Store product at –70°C. For optimal storage, aliquot target into smaller quantities after centrifugation and store at recommended temperature. For most favorable performance, avoid repeated handling and multiple freeze/thaw cycles. |
| Publications : |
| Gene Name | PTPN11 protein tyrosine phosphatase, non-receptor type 11 [ Homo sapiens ] |
| Synonyms | protein tyrosine phosphatase, non-receptor type 11; CFC; NS1; SHP2; BPTP3; PTP2C; PTP-1D; SH-PTP2; SH-PTP3; MGC14433; PTPN11; protein tyrosine phosphatase-2; protein-tyrosine phosphatase 2C; EC 3.1.3.48; PTP-2C; SHP-2; SHPTP2; Shp2; Noonan syndrome 1; Protein-tyrosine phosphatase 2C |
| Gene ID | 5781 |
| mRNA Refseq | NM_002834 |
| Protein Refseq | NP_002825 |
| MIM | 176876 |
| UniProt ID | Q06124 |
| Chromosome Location | 12q24 |
| Pathway | Adipocytokine signaling pathway; Chronic myeloid leukemia; Epithelial cell signaling in Helicobacter pylori infection; Jak-STAT signaling pathway; Leukocyte transendothelial migration; Natural killer cell mediated cytotoxicity; Neurotrophin signaling pathway; Renal cell carcinoma |
| Function | hydrolase activity; insulin receptor binding; insulin receptor substrate binding; non-membrane spanning protein tyrosine phosphatase activity; peptide hormone receptor binding; phospholipase binding; protein domain specific binding |
| ◆ Recombinant Proteins | ||
| PTPN11-04H | Active Recombinant Human PTPN11 Protein(Full length), N-GST-tagged | +Inquiry |
| PTPN11-6745H | Recombinant Human PTPN11 protein, His-tagged | +Inquiry |
| PTPN11-6100H | Recombinant Full Length Human PTPN11 Protein (Met1-Arg593), N-His tagged | +Inquiry |
| PTPN11-18H | Active Recombinant Human PTPN11 Protein, N-terminal 6XHis and AviTag™, Biotinylated | +Inquiry |
| PTPN11-31402TH | Active Recombinant Human PTPN11 protein, GST-tagged | +Inquiry |
| ◆ Cell & Tissue Lysates | ||
| PTPN11-1334MCL | Recombinant Mouse PTPN11 cell lysate | +Inquiry |
| PTPN11-283HKCL | Human PTPN11 Knockdown Cell Lysate | +Inquiry |
Decline in arylsulfatase B expression increases EGFR expression by inhibiting the protein-tyrosine phosphatase SHP2 and activating JNK in prostate cells
Journal: The Journal of Biological Chemistry PubMed ID: 29794138 Data: 2018/7/13
Authors: Sumit Bhattacharyya, Leo Feferman, Joanne K. Tobacman
Article Snippet:Then preparations were blocked for 1 h at RT with blocking buffer (PBS, pH 7.4, with 1% BSA).Then preparations were blocked for 1 h at RT with blocking buffer (PBS, pH 7.4, with 1% BSA).. After blocking, 100 μl of (0.5 μg/ml) PTPN11-GST (Creative BioMart, Shirley, NY) in PBS was added to each well.. Plates were incubated at RT for 2 h and then washed three times with wash buffer.Plates were incubated at RT for 2 h and then washed three times with wash buffer.
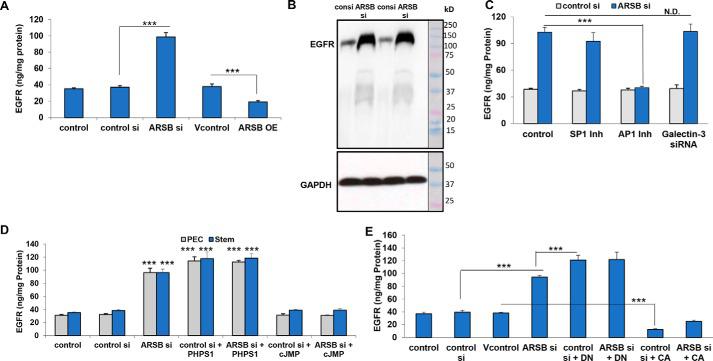
EGFR is increased following decline in ARB or
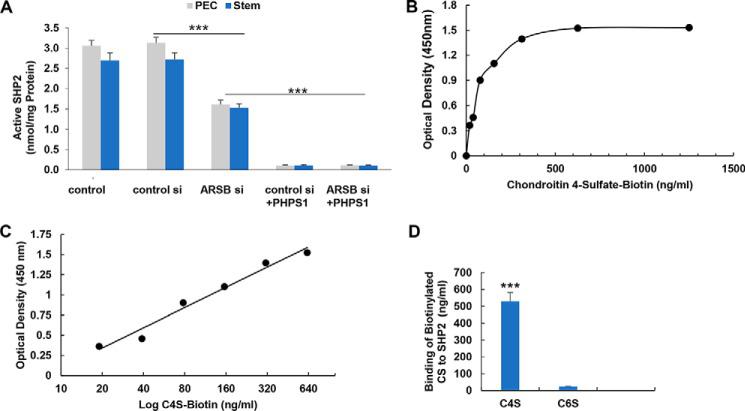
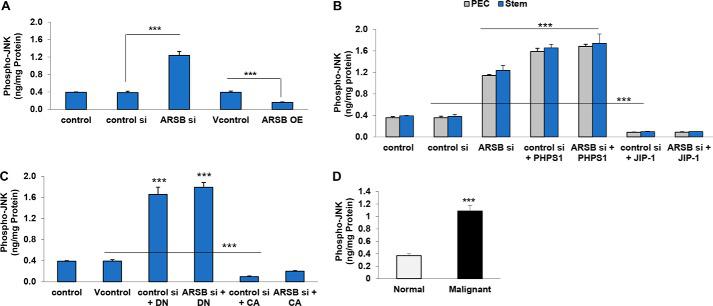
Phospho-JNK is increased in prostate stem cells and prostate tissue following ARSB silencing or
Not For Human Consumption!
Inquiry
- Reviews (0)
- Q&As (0)

Ask a Question for All PTPN11 Products
Required fields are marked with *
My Review for All PTPN11 Products
Required fields are marked with *