
Immunoglobulin Superfamily CAMs
Related Symbol Search List
- ALCAM
- BCAM
- BVES
- CADM1
- CADM2
- CADM3
- CADM4
- CD160
- CD47
- CDO1
- CEACAM1
- CEACAM16
- CEACAM18
- CEACAM19
- CEACAM20
- CEACAM21
- CEACAM3
- CEACAM4
- CEACAM5
- CEACAM6
- CEACAM7
- CEACAM8
- ASAM
- CNTN1
- CNTN3
- CNTN4
- CNTN5
- DDX11
- DSCAM
- ESAM
- F11R
- HEPACAM
- ICAM-1
- ICAM-2
- CD50
- ICAM4
- ICAM5
- IGDCC3
- IGSF11
- IGSF21
- IGSF3
- IGSF5
- ISLR2
- ITGB1BP1
- Jam3
- Jam2
- Jam3
- JAML
- KIRREL
- KIRREL2
- KIRREL3
- L1CAM
- LAMP3
- LRFN1
- LRFN2
- LRFN3
- LRFN4
- LRFN5
- LSAMP
- MADCAM1
- MCAM
- MDGA1
- MDGA2
- MFGE8
- MXRA8
- NCAM1
- NCAM2
- Pvrl2
- NEGR1
- NFASC
- NINJ1
- NINJ2
- Nephrin
- NPHS2
- NPTN
- OPCML
- PEAR1
- CD31
- SIRPA
- TROP2
- THBS1
- TMEM204
- CD28H
- TMIGD2
- UBE2S
- VCAM1
Immunology Background
Cell Adhesion Molecules (CAMs) are subdivided into four families such as selectins and their mucin family ligands, integrins and immunoglobulin superfamily (IgSF) molecules, which mediate different steps in leukocyte adhesion and emigration.
Selectins
The selectins consist of three different glycoproteins which share the common presence of an NH2-terminal C-type (Ca2+ dependent) lectin binding domain, an epidermal growth factor like region (EGF) and a variable number of short consensus repeat motifs (SCR) which are related to those in complement regulatory proteins. The variability in number of SCR motifs ultimately leads to different physical lengths of the selectin protein which likely functions to place the lectin binding domain of the selectins at varying distances from the cell surface. All types of currently described selectins also share homologous transmembrane spanning regions and contain a short intra-cytoplasmic region which, like intra-cytoplasmic regions of other CAMs, interacts with the microfilament component of the cytoskeleton and function in cellular signaling including activation. The ligand binding activity of selectins is primarily at the lectin-binding domain with a variable contribution from EGF and SCR domains. Selectin-ligand interactions are responsible for the initial contact of leukocytes with the vascular endothelium.
There are three subsets of selectins:
E-selectin, also called antigen-like family member E (CD62E), endothelial-leukocyte adhesion molecule 1 (ELAM-1), or leukocyte-endothelial cell adhesion molecule 2 (LECAM2), is a selectin cell adhesion molecule which only expressed on endothelial cells activated by cytokines. Like other selectins, it plays a vital part in inflammation. In humans, E-selectin is encoded by the SELE gene.
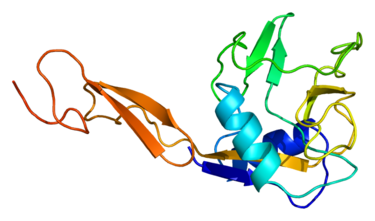 Figure 1. Structure of the SELE protein. Based on PyMOL rendering of PDB 1esl.
Figure 1. Structure of the SELE protein. Based on PyMOL rendering of PDB 1esl.
L-selectin, also called CD62L, is a cell adhesion molecule found on leukocytes and the preimplantation embryo. It belongs to the selectin family of proteins, which recognize sialylated carbohydrate groups. It is cleaved by ADAM17. SELL is a cell surface component that is a member of a family of adhesion/homing receptors that play important roles in lymphocyte-endothelial cell interactions. The molecule is composed of multiple domains: one homologous to lectins, one to epidermal growth factor, and two to the consensus repeat units found in C3/C4-binding proteins.
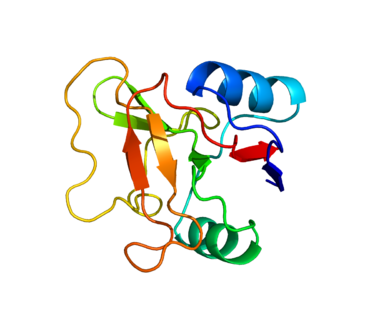 Figure 2. Structure of protein SELL.Based on PyMOL rendering of PDB 1KJB.
Figure 2. Structure of protein SELL.Based on PyMOL rendering of PDB 1KJB.
P-selectin is a protein that in humans is encoded by the SELP gene. P-selectin functions as a cell adhesion molecule (CAM) on the surfaces of activated endothelial cells, which line the inner surface of blood vessels, and activated platelets. In unactivated endothelial cells, it is stored in granules called Weibel-Palade bodies. In unactivated platelets P-selectin is stored in α-granules. Other names for P-selectin include CD62P, Granule Membrane Protein 140 (GMP-140), and Platelet Activation-Dependent Granule to External Membrane Protein (PADGEM). It was first identified in endothelial cells in 1989.
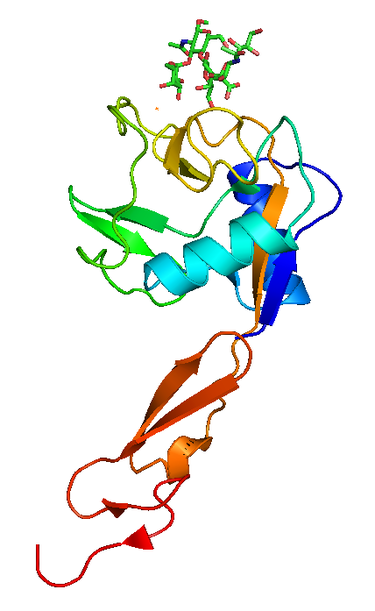 Figure 3. P selectin lectin, PDB 1G1R
Figure 3. P selectin lectin, PDB 1G1R
Integrin
Integrins, one of the major classes of receptors within the ECM, mediates cell–ECM interactions with collagen, fibrinogen, fibronectin, and vitronectin. Integrins provide essential links between the extracellular environment and the intracellular signalling pathways, which can play roles in cell behaviours such as apoptosis, differentiation, survival, and transcription. Integrins are heterodimeric, as they consist of an alpha and beta subunit. There are currently 18 alpha subunits and 8 beta subunits, which combine to make up 24 different integrin combinations. Within each of the alpha and beta subunits there is a large extracellular domain, a transmembrane domain and a short cytoplasmic domain. The extracellular domain is where the ligand binds through the use of divalent cations. In general, Mn2+ increases affinity, Mg2+ promotes adhesion to cells, and Ca2+ decreases cell adhesion. Integrins regulate their activity within the body by changing conformation. Most exist at rest in a low affinity state, which can be altered to high affinity through an external agonist which causes a conformational change within the integrin, increasing their affinity. An example of this is the aggregation of platelets;[9] Agonists such as thrombin or collagen trigger the integrin into its high affinity state, which causes increased fibrinogen binding, causing platelet aggregation.
Immunoglobulin Superfamily Molecules
The immunoglobulin superfamily (IgSF) is a large protein superfamily of cell surface and soluble proteins that are involved in the recognition, binding, or adhesion processes of cells. Molecules are categorized as members of this superfamily based on shared structural features with immunoglobulins (also known as antibodies); they all possess a domain known as an immunoglobulin domain or fold. Members of the IgSF include cell surface antigen receptors, co-receptors and co-stimulatory molecules of the immune system, molecules involved in antigen presentation to lymphocytes, cell adhesion molecules, certain cytokine receptors and intracellular muscle proteins. They are commonly associated with roles in the immune system. Otherwise, the sperm-specific protein IZUMO1, a member of the immunoglobulin superfamily, has also been identified as the only sperm membrane protein essential for sperm-egg fusion.
Cell Adhesion Molecules in the Leukocyte Adhesion Cascade
The recruitment of leukocytes from the blood stream to extravascular sites of inflammation is necessary for an effective inflammatory response. The recruitment is mediated by Cell Adhesion Molecules, cytokines and chemoattractants which together coordinate this complex series of events. The adhesion cascade involves five steps which occur in sequence but may overlap in time. At sites of inflammation in the systemic circulation (as opposed to the pulmonary circulation), post-capillary venule endothelium becomes activated by the local generation of a variety of inflammatory mediators. Histamine, thrombin and LTC4 lead to a rapid expression of P-selectin on the endothelial surface while the early proinflammatory cytokines such as IL-1 and TNFα lead to de novo synthesis of both P- and E-selectin as well as ICAM-1 and VCAM-1 resulting in increased surface expression at time points greater than 2 hours following stimulation. When endothelium is exposed to these cytokines especially in the presence of FN for prolonged periods (as during chronic infection), the volume of the endothelial cell expands and it becomes columnar, similar to the morphology in high endothelial venules in lymph nodes.
References:
1. Korthuis RJ.; et al. Role of neutrophil-endothelial cell adhesion in inflammatory disorders. J Crit Care. 1994, 9 (1): 47-71.
2. Chothia C.; et al. The molecular structure of cell adhesion molecules. Annu. Rev. Biochem. 1997,66: 823-62.